Marfanův syndrom
 Marfanův syndrom je nazýván patologie charakterizované zaostalost pojivové tkáně u dětí v embryonální nebo postnatálním období, v důsledku strukturních defektů v kolagenu, a je doprovázen lézí především pohybového systému, kardiovaskulárního systému a očí.
Marfanův syndrom je nazýván patologie charakterizované zaostalost pojivové tkáně u dětí v embryonální nebo postnatálním období, v důsledku strukturních defektů v kolagenu, a je doprovázen lézí především pohybového systému, kardiovaskulárního systému a očí.
Obsah
Podle různých autorů, je výskyt tohoto onemocnění je 1 na každých 10-20 tisíc. Novorozenců bez pohlaví nebo rasy.
patologie Příčiny
Marfanův syndrom - vrozená malformace, dědí autosomálně dominantním způsobem. Na srdeční onemocnění je mutace v FBN1 genu, který je zodpovědný za syntézu fibrillin - strukturní protein extracelulární matrix je důležitá sloučenina, která poskytuje pružnost a kontraktilitu pojivové tkáně.
Video: Marfanův syndrom
To znamená, že hlavní příčinou Marfanův syndrom je nedostatek fibrillin, což vede k narušení formování vláknité struktury, ztráta pojivové tkáně pružnosti a pevnosti, a, jako výsledek, neschopnost odolávat fyziologické zátěže.
Přibližně 75% případů u rodinného dědictví tohoto syndromu zjištěných ve zbývajících 25% - primární mutace.
Podle výzkumu, věk čím více, tím otce (obzvláště po věk 35), tím vyšší je riziko, že dítě s Marfanův syndrom.
klasifikace nemocí
Marfanův syndrom je zařazen do několika typů v závislosti na počtu postižených systémech u člověka:
Video: Marfanův syndrom
- Vymazány tvar vyznačující se tím, mírnými změnami v jedné nebo dvou systémů;
- Vyjádřeno forma, která je charakterizována změnami v alespoň jednom sisteme- abnormalit ve dvou nebo více sistemah- mírnými postižením ve třech systémech.
Podle charakteru proudění je rozděleno do stabilní syndromu a progresivní.
Příznaky Marfanův syndrom
Klinický obraz tohoto onemocnění charakterizované destrukcí mnoha a různých životně důležitých orgánů a systémů těla: skeletu, očí, nervového a kardiovaskulárního systému.
Ve většině případů, symptomy Marfanův syndrom, jsou:
- Vysoký růst;
- Relativně krátký trup;
- Neúměrně dlouhé a tenké končetiny (dolichostenomelia);
- Pavoukovci protáhlé prsty (arachnodactyly);
- Astenická ústava;
- hypotonie;
- Špatně vyvinutý podkožní tuk;
- Dlouhé a úzké obličejového skeletu (dolichocephaly);
- Vysoká obloukovité patro;
- Malocclusion.
Průměrná délka těla u dívek s Marfanův syndrom je 52,5 cm při narození, konečná výška - 175 cm, v chlapců - 53 a 191 cm, v tomto pořadí.
Také onemocnění charakterizované abnormalitami v kloubech (hypermobility), deformity páteře (kyfóza, podvrtnutí a subluxace krční páteře, skolióza, spondylolistéza, kyfoskolióza), výstupek acetabula, ploché nohy.
Klinický obraz syndromu dominuje kardiovaskulární onemocnění, které se často určuje výsledek. Zjevné vady příčky a valvulární nemoc srdce, defekty ve struktuře stěn nádoby, zejména hlavní větve plicní tepně a aorty a vrozených srdečních vad: plicní stenóza, atriální defekt a komorového septa, aorty koarktace, atd.
Nejnepříznivější forma Marfanův syndrom, což se projevuje při narození v klasické verzi, vede k progresivnímu selhání srdce, a v důsledku toho k smrti v průběhu prvního roku života.
VIDEO: Top 10 nejstrašidelnější genetické abnormality
Ve většině případů, Marfanův syndrom je příznakem a patologie orgánu zraku, vyznačující se krátkozrakost, šilhání, hypoplazie ciliární sval a duhovky, zvýšení velikosti a zploštění rohovky, ektopie čočky, retinální cévní variace ráže.
Také u dětí s touto nemocí diagnostikováno poškození nervové a bronchopulmonální systému a dalších orgánů: ledvin ektopie, přestávek a podvrtnutí vazů, křečové žíly, stehenní a tříselné kýla, prolaps močového měchýře a dělohy u dívek, kožních lézí a měkké tkáně.
Marfanův syndrom je charakterizován vysokou adrenalinu, který je důvod, hyperaktivita, nervózní vzrušení, a někdy i duševní dotace a mimořádné vývojové schopnosti.
Diagnostika Marfanův syndrom
Konečná diagnóza může být provedena až po komplexní vyšetření pacienta, který se zúčastnil genetik, ortoped, kardiolog a očního lékaře.
 Cílem genetiky je studie o rodinnou historii a včetně identifikace příbuzných, kteří zemřeli na kardiovaskulární onemocnění. Za účelem diagnózy Marfanův syndrom kardiologem přiřadí echokardiogram, EKG, rentgen hrudníku. Oční lékař zkoumá oko s štěrbinovou lampou pro detekci posunutí čočky nebo jiných patologických stavů. Ortoped prozkoumá hrudník a páteř v přítomnosti skoliózy, ploché nohy, atd.
Cílem genetiky je studie o rodinnou historii a včetně identifikace příbuzných, kteří zemřeli na kardiovaskulární onemocnění. Za účelem diagnózy Marfanův syndrom kardiologem přiřadí echokardiogram, EKG, rentgen hrudníku. Oční lékař zkoumá oko s štěrbinovou lampou pro detekci posunutí čočky nebo jiných patologických stavů. Ortoped prozkoumá hrudník a páteř v přítomnosti skoliózy, ploché nohy, atd.
Pokud se potvrdí verze dědičnosti, pro definitivní diagnózu vyžaduje přítomnost charakteristických rysů Marfanův syndrom v alespoň dvou systémech těla. V případě, že žádné dědictví, je nutno potvrdit porážku nejméně tří tělesných systémů.
Marfanův syndrom Léčba
Některé způsoby léčení Marfanův syndrom, bohužel, neexistuje. Hlavním cílem léčby - odstranění komorbidit a prevence komplikací.
Léčba progresivní skoliózy provádí fyzioterapii a mechanické upevnění skeletu, operační korekce se provádí v některých případech.
Někteří pacienti ukazují chirurgickou plastickou aortu, aortální a mitrální ventily.
korekce se provádí pomocí brýlí nebo čoček, léčba se podává podle potřeby šedý zákal, glaukom atd.
Tak, všichni pacienti vykazovaly symptomatickou terapii.
 Frederick syndrom: příčiny, příznaky, léčba
Frederick syndrom: příčiny, příznaky, léčba Rhabdomyomas srdce u novorozenců
Rhabdomyomas srdce u novorozenců Meningeální syndrom - příčiny, příznaky, léčba
Meningeální syndrom - příčiny, příznaky, léčba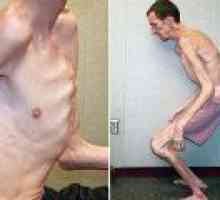 Marfanův syndrom - příčiny, příznaky, léčba
Marfanův syndrom - příčiny, příznaky, léčba Patau syndrom - Způsobuje prognóza, léčba
Patau syndrom - Způsobuje prognóza, léčba Werner syndrom - existuje cesta ven?
Werner syndrom - existuje cesta ven? Gilbertův syndrom - příčiny, příznaky, léčba
Gilbertův syndrom - příčiny, příznaky, léčba Albinismus
Albinismus Klinefelterův syndrom
Klinefelterův syndrom Mukopolysacharidóza
Mukopolysacharidóza Sítnice disinsertion
Sítnice disinsertion Syndrom Dandy-Walker
Syndrom Dandy-Walker Patau syndrom
Patau syndrom Williams syndrom
Williams syndrom Gilbertův syndrom
Gilbertův syndrom Rozedma plic - příznaky, léčba
Rozedma plic - příznaky, léčba Tkáň onemocnění - příčiny, diagnostika, léčba
Tkáň onemocnění - příčiny, diagnostika, léčba Jedenáct Pronatal - návod k použití, real
Jedenáct Pronatal - návod k použití, real Příznaky a léčba pojivové tkáně dysplazie
Příznaky a léčba pojivové tkáně dysplazie Příznaky a léčení syndromu suchého oka
Příznaky a léčení syndromu suchého oka Edwards syndrom: způsobuje patologii
Edwards syndrom: způsobuje patologii